

Understanding alpha-1
What is alpha1-antitrypsin (AAT) deficiency (alpha-1)?
Alpha-1, the major known genetic risk factor for chronic obstructive pulmonary disease (COPD), is one of the world's most prevalent, potentially fatal hereditary diseases.1,2
The genetics behind alpha-13,4
AAT deficiency is an inherited condition that is caused by mutations in the SERPINA1 gene, which is responsible for producing the AAT protein. The AAT protein, which is produced in the liver and then released in the blood, is responsible for protecting the body from the neutrophil elastase enzyme.3
When mutations occur in the SERPINA1 gene, the result is the production of an abnormal protein that gets trapped in the liver, resulting in low serum levels of AAT. Low serum levels of AAT can predispose to lung breakdown by neutrophil elastase and other enzymes that break down proteins.3
To date, more than 150 different mutations have been identified in the SERPINA1 gene.
The most common mutations are the S and Z alleles, which cause AAT deficiency3:
- The Z allele is associated with very low serum levels of AAT (~10%-15% of normal levels)
- The S allele is associated with moderately low serum levels of AAT
The M allele is the most common and produces normal levels of the ATT protein.3,4
AAT deficiency is a codominant genetic condition. Codominant genetic conditions occur when each inherited allele expresses some effect, such as lowering serum levels of AAT. Generally, in a codominant condition, an individual who inherits 2 copies of a mutated gene (ZZ) has a higher risk of developing emphysema than someone inheriting 1 abnormal allele (MZ; low risk of developing disease, only in the absence of smoking).3
What is the risk of passing on alpha-1?3
The chance that 2 carrier parents will pass on the mutated gene and have an affected (ZZ) child
The chance that a child of 2 carrier parents will become a carrier
The chance that a child of 2 carrier parents will receive normal genes
In the case of an individual inheriting an S allele and a Z allele (SZ), the risk of developing chronic obstructive pulmonary disease is increased if he or she smokes.
Because AAT is an autosomal condition, the risk of inheriting a defective gene is the same for both males and females because the gene is not on the sex chromosome (X or Y).
Symptoms of lung disease associated with AAT5
For patients with alpha-1, the most common symptoms of lung disease include:
- Shortness of breath
- Wheezing
- Chronic bronchitis (cough and sputum production)
- Recurring chest colds
- Low tolerance for exercise
- Year-round allergies
- Bronchiectasis
Early diagnosis and treatment of alpha-1 is very important. However, alpha-1 can't be diagnosed by symptoms or by a medical examination alone; patients need to be screened to know for sure.6
Symptoms are similar to COPD
COPD describes multiple lung diseases, including emphysema and chronic bronchitis. Alpha-1 often goes undiagnosed because its symptoms are similar to COPD.1,4
Symptoms alone cannot identify a patient with alpha-1
It's critical to remember that a patient with alpha-1 cannot be identified by symptoms or medical examination alone. The only way to truly know if a patient has alpha-1 is through a laboratory test.6
Alpha-1 can be found in patients of any age6,7
While the symptoms of alpha-1 usually appear between the ages of 20 and 50 years of age, data obtained over a 3-year period (2004–2007) from the renowned Alpha-1 Genetics Laboratory in Florida showed that the majority of newly diagnosed patients were 50 years of age or older.6,7
Smokers and ex-smokers make up a large part of the diagnosed alpha-1 population
Smoking accelerates lung function decline in patients with alpha-1. In a national registry study of 1129 patients with alpha-1, approximately 80% were either current smokers (8%) or ex-smokers (71%).8,9
Given the disparity in patient diagnoses, ALL patients with COPD should be screened for alpha-1, regardless of smoking history.10
FEV1 levels alone should not determine whom to screen
These readings can be misleading because some nonsmokers have low FEV1 levels and some smokers have high FEV1 levels.8
FEV1=forced expiratory volume in 1 second.
AAT deficiency can lead to serious lung destruction
AAT is a protein with potent protease inhibitor activity. Its main function is to protect normal lung tissue from proteolytic attack during inflammation, such as that caused by infection and inhaled irritants like tobacco smoke. When low levels of AAT occur, the lungs are at risk for serious lung disease.11-14
Low levels of circulating AAT allow potentially harmful enzymes, like neutrophil elastase, to remain in the lungs unchecked. This combination of low levels of AAT and the consequent proliferation of neutrophil elastase leave lung tissue vulnerable to destruction, resulting in a decline in lung function. Alpha-1 may be a contributing cause of up to 3% of all COPD cases in the United States.1,11,12
The significance of AAT
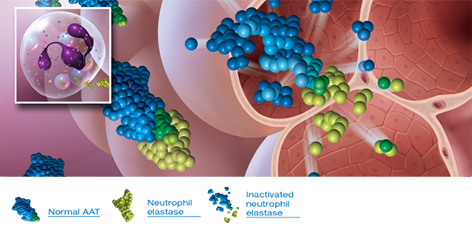
Endogenous AAT is critical to safeguarding the lungs11
Lungs with normal AAT levels keep neutrophil elastase in check so that lung structure is preserved11
- Neutrophil elastase is released in response to lung infection, injury, or inflammation2
- Produced in the liver, AAT inhibits excess amounts of proteases11
- Only laboratory testing can determine if an individual's AAT levels are sufficient6
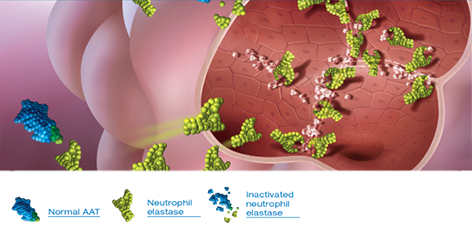
Low levels of AAT leave lung tissue vulnerable to destruction11
In AAT-deficient individuals, excess neutrophil elastase cannot be neutralized, lung elastin is destroyed, and lung function is compromised11
- Smoking, secondary smoke, dust, and exposure to fumes accelerate lung disease in patients with alpha-111,15
- Bacterial infections are additional risk factors for lung disease in patients with alpha-111
Major clinical guidelines, including the American Thoracic Society, recommend screening all patients with COPD for alpha-1, regardless of age, smoking history, and FEV1 decline1,10,15
Resources for you and your patients
To help your practice and your patients learn more about alpha-1, access such resources as videos, peer testimonials, and downloadable PDFs.

IMPORTANT SAFETY INFORMATION
PROLASTIN®-C LIQUID is an alpha1-proteinase inhibitor (human) (alpha1-PI) indicated for chronic augmentation and maintenance therapy in adults with clinical evidence of emphysema due to severe hereditary deficiency of alpha1-PI (alpha1-antitrypsin deficiency).
Limitations of Use
- The effect of augmentation therapy with any alpha1-PI, including PROLASTIN-C LIQUID, on pulmonary exacerbations and on the progression of emphysema in alpha1-PI deficiency has not been conclusively demonstrated in randomized, controlled clinical trials
- Clinical data demonstrating the long-term effects of chronic augmentation or maintenance therapy with PROLASTIN-C LIQUID are not available
- PROLASTIN-C LIQUID is not indicated as therapy for lung disease in patients in whom severe alpha1-PI deficiency has not been established
PROLASTIN-C LIQUID is contraindicated in immunoglobulin A (IgA)-deficient patients with antibodies against IgA or patients with a history of anaphylaxis or other severe systemic reaction to alpha1-PI products.
Hypersensitivity reactions, including anaphylaxis, may occur. Monitor vital signs and observe the patient carefully throughout the infusion. If hypersensitivity symptoms occur, promptly stop PROLASTIN-C LIQUID infusion and begin appropriate therapy.
Because PROLASTIN-C LIQUID is made from human plasma, it may carry a risk of transmitting infectious agents, eg, viruses, the variant Creutzfeldt-Jakob disease (vCJD) agent, and, theoretically, the Creutzfeldt-Jakob disease (CJD) agent. This also applies to unknown or emerging viruses and other pathogens.
The most common adverse reactions during PROLASTIN-C LIQUID clinical trials in >5% of subjects were diarrhea and fatigue, each of which occurred in 2 subjects (6%).
Please see full Prescribing Information for PROLASTIN-C LIQUID.
You are encouraged to report negative side effects of prescription drugs to the FDA. Visit www.fda.gov/medwatch or call 1-800-FDA-1088.
References:
1. World Health Organization. α1-antitrypsin deficiency: memorandum from a WHO meeting. Bull World Health Organ. 1997;75(5):397-415. 2. de Serres FJ. Alpha-1 antitrypsin deficiency is not a rare disease but a disease that is rarely diagnosed. Environ Health Perspect. 2003;111(16):1851-1854. 3. National Organization for Rare Disorders. Alpha-1 antitrypsin deficiency. https://rarediseases.org/rare-diseases/alpha-1-antitrypsin-deficiency/. Accessed September 25, 2018. 4. National Human Genome Research Institute. Learning about alpha-1 antitrypsin deficiency (AATD). https://www.genome.gov/19518992